

A Reliable and Efficient Resonance Theory Based on Analysis of DFT Wave Functions
发布时间:2021-03-21
点击次数:
DOI码:10.1039/D0CP06207C
所属单位:扬州大学
发表刊物:Physical Chemistry Chemical Physics
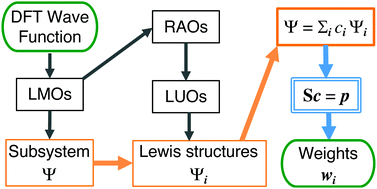
摘要:Due to methodological difficulties and limitations of applicability, a quantitative bonding analysis based on the theory of resonance is presently not as convenient and popular as that based on the molecular orbital (MO) methods. Here, we propose an efficient quantitative resonance theory by expanding the DFT wave function in terms of a complete set of Lewis structures. By rigorously separating the resonance subsystem represented by a set of localized MOs, this approach is able to treat large molecules, nonplanar π-conjugate systems, and bonding systems mixing both σ and π electrons. Assessment in 2c-2e systems suggests a new projection-weighted symmetric orthogonalization method to evaluate the weights of resonance contributors, which overcomes the drawbacks of other weighting schemes. Applications to benzene, naphthalene and chlorobenzene show that the present method is insensitive to the basis set employed in the DFT calculations, and to the choices of the independent Lewis set determined by Rumer's rule. Advanced applications to diverse chemical problems provide unique and valuable insights into the understanding of hydrogen bonding, the π substituent effect on benzene, and the mechanism of Diels–Alder reactions.
第一作者:汪洋
论文类型:基础研究
学科门类:理学
文献类型:SCI
卷号:23
页面范围:2331-2348
是否译文:否
发表时间:2020-12-18
收录刊物:SCI
发布期刊链接:https://pubs.rsc.org/en/content/articlelanding/2021/cp/d0cp06207c